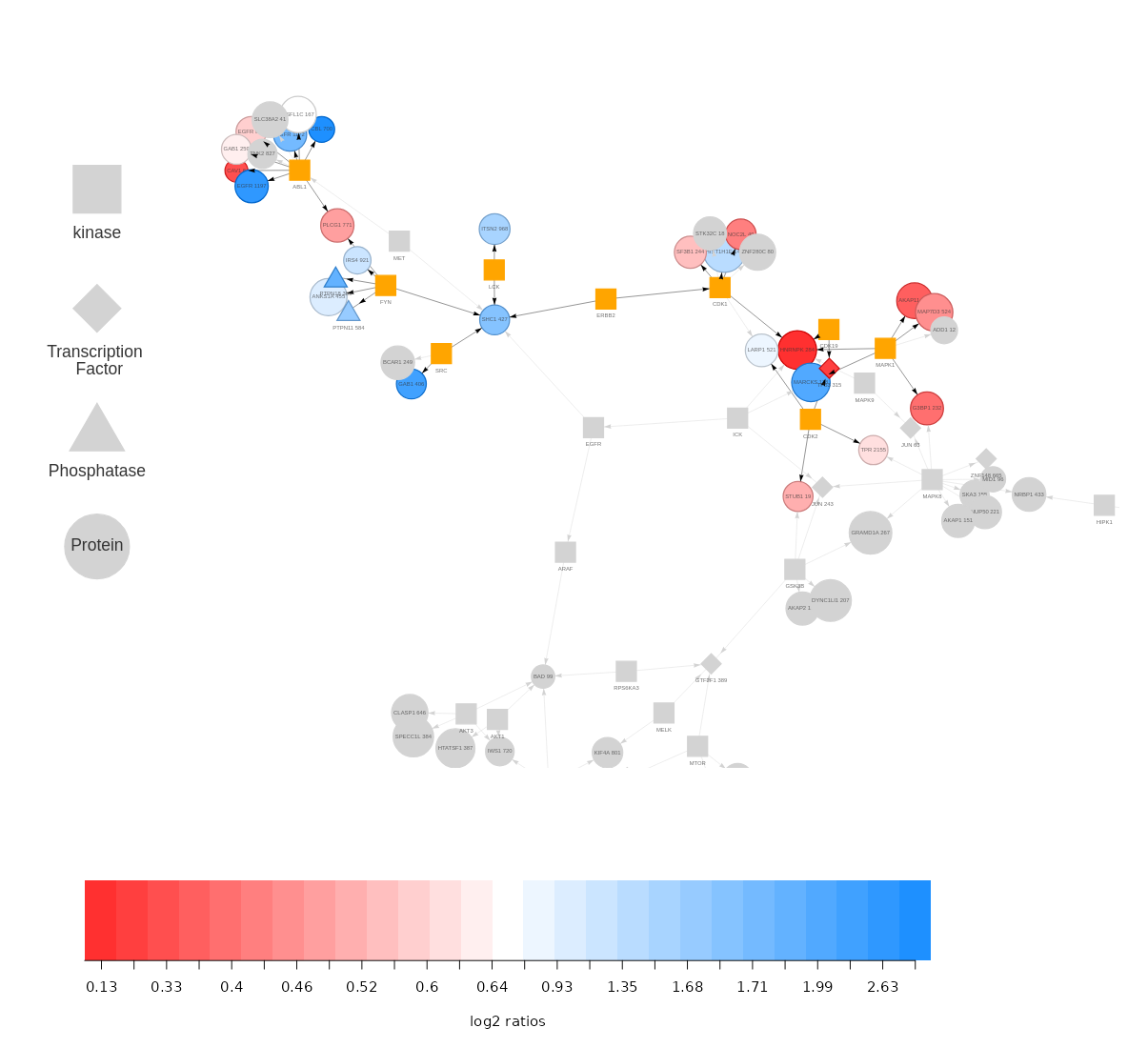
Welcome to the SELPHI2 server
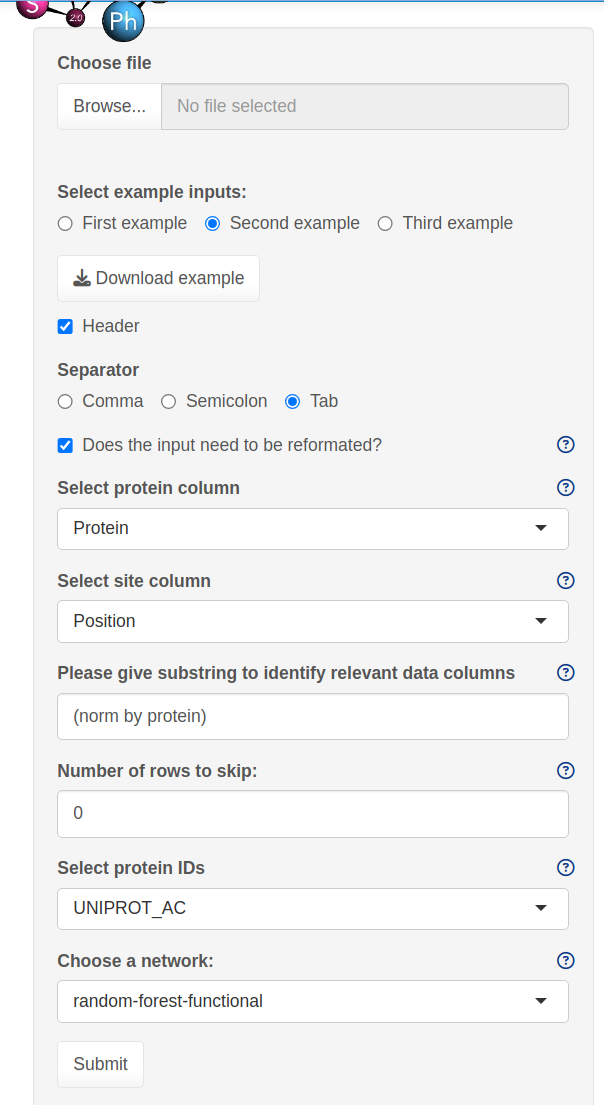
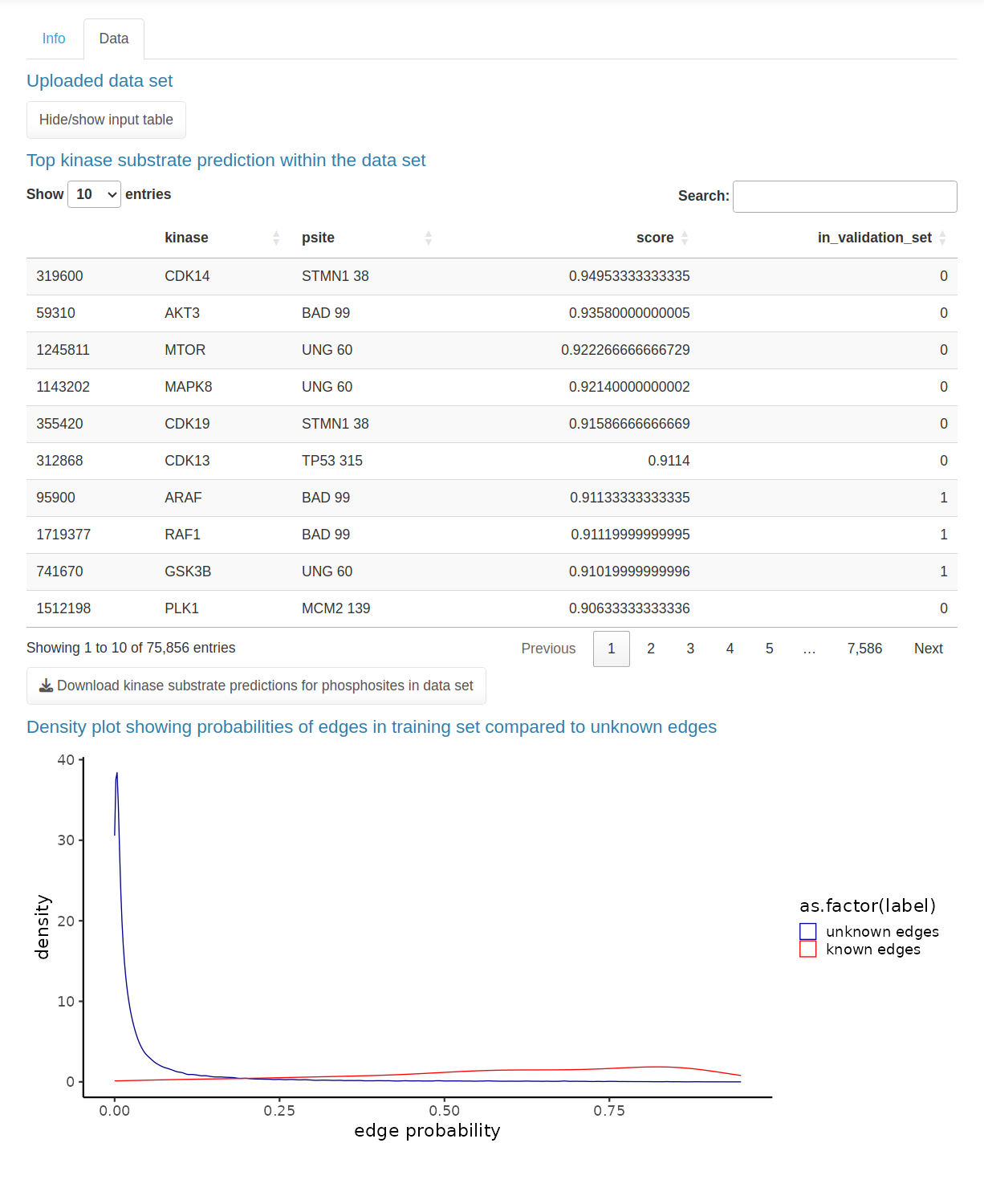
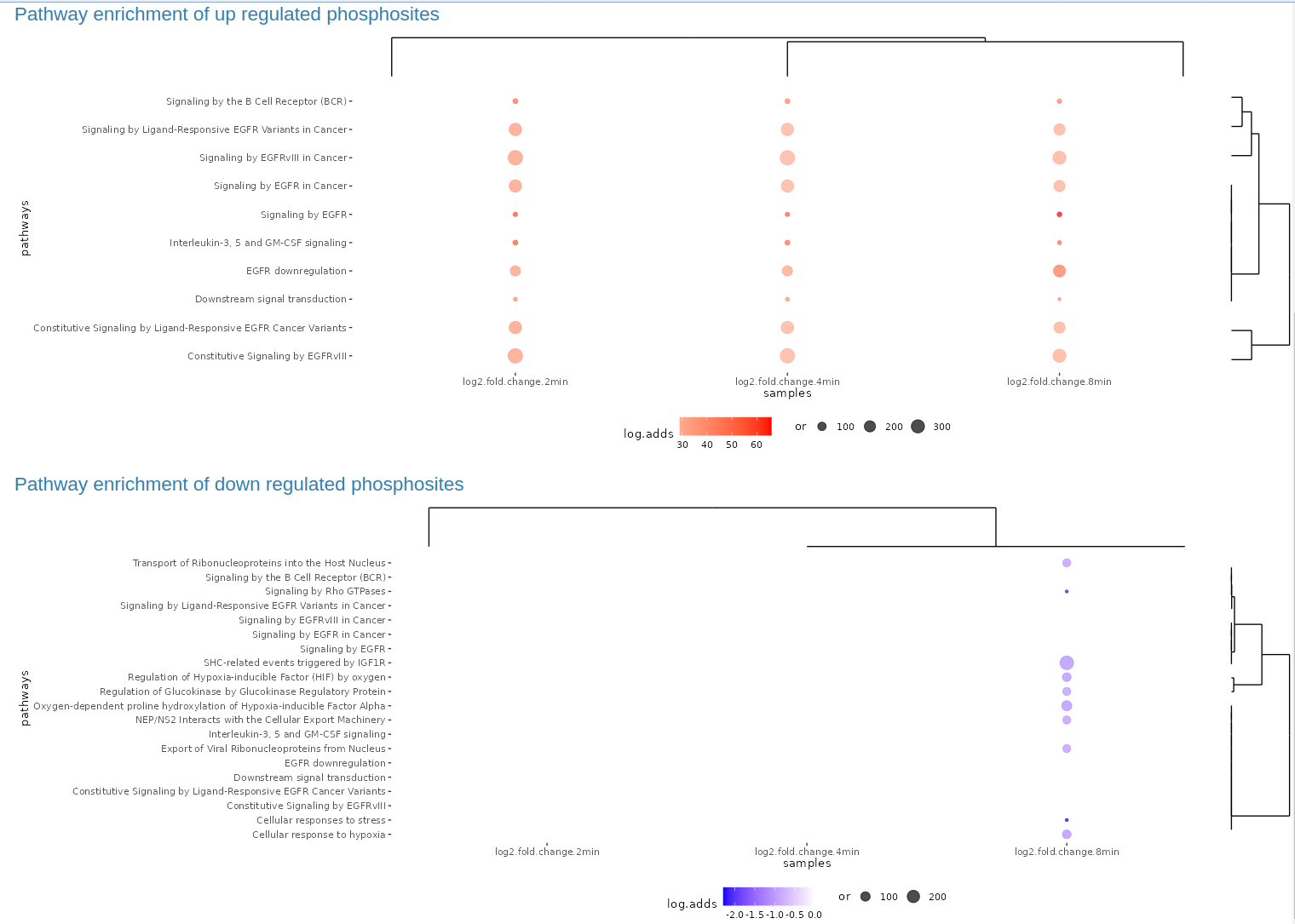
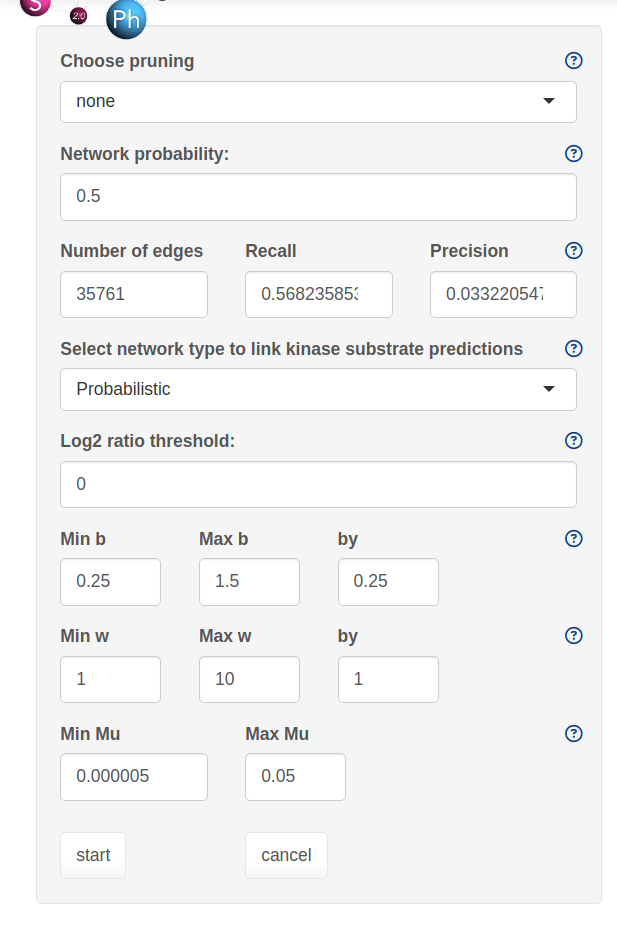
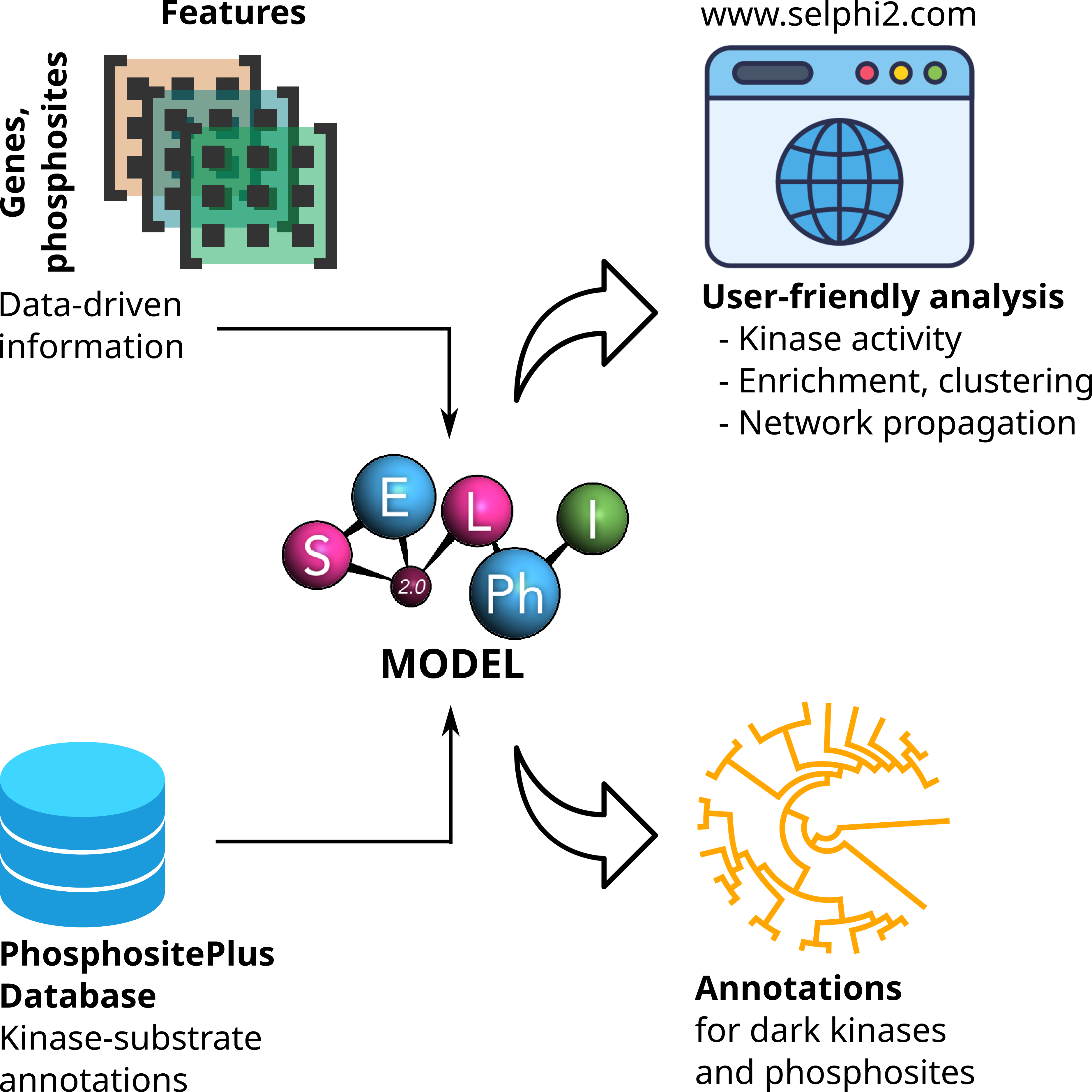
The SELPHI 2.0 (Systematic Extraction of Linked PhosphoInteractions 2.0) server provides you with a platform to analyse phosphoproteomics data, including providing you with a list of high confidence kinase-substrate predictions for the phosphosites included in their data. SELPHI 2 contains 73+ million kinase substrate predictions. You can also fit the kinase substrate predictions to their data set to identify context-specific sub networks, conduct pathway enrichments and download highly probable edges supported by external evidence from external publications.
If you find SELPHI2.0 useful please cite:
Maier BD#, Petursson B#, Lussana A# and Petsalaki E, SELPHI 2: Data-driven extraction of human kinase-substrate relationships from omics datasets. bioRxiv, 2024. https://doi.org/10.1101/2022.01.15.476449
(#) These authors contributed equally.
All code for reproducing this project as well as a Docker image of the web server can be found at: https://gitlab.ebi.ac.uk/petsalakilab/selphi_2. The feature and prediction matrix can be downloaded from Zenodo.
License for SELPHI2 server
Copyright (c) 2024 Petsalaki Group, EMBL-EBI
Permission is hereby granted, free of charge, to any person obtaining a copy of this software and associated documentation files (the "Software"), to deal in the Software without restriction, including without limitation the rights to use, copy, modify, merge, publish, distribute, sublicense, and/or sell copies of the Software, and to permit persons to whom the Software is furnished to do so, subject to the following conditions:The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software.
THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.